SRA(Sequence ReadArchive)資料庫:
是NCBI(National Center for Biotechnology Information)旗下用於存儲高通量定序資料的資料庫,在此處都可以免費下載所有已發表的文獻中的高通量定序數據基本都上傳至此,方便其他研究者下載及再研究。
其中的數據則是通過壓縮後以.sra文件格式來保存的,SRA資料庫可以用於搜索和展示SRA項目數據,包括SRA首頁和Entrez system,由NCBI 負責維護。
ENA(European Nucleotide Archive) 資料庫:
屬EBI (European Bioinformatics Institute),功能等同SRA,並且對保存的數據做了註釋,對於有數據需求的研究人員來說,ENA資料庫最誘人的點應該是可以直接下載fastq (.gz)文件,由EBI 負責維護。
兩者在主要功能方面非常類似,同時數據互通。
本文主要是從NCBI 上的SRA 資料庫中聊聊如何下載我們想要的fastq 檔(raw data)。
下載數據之前,先聊聊SRA資料庫的一些基礎知識。 SRA資料庫的組織框架是基於 STUDY, SAMPLE, EXPERIMENT, RUN 四個概念構建的
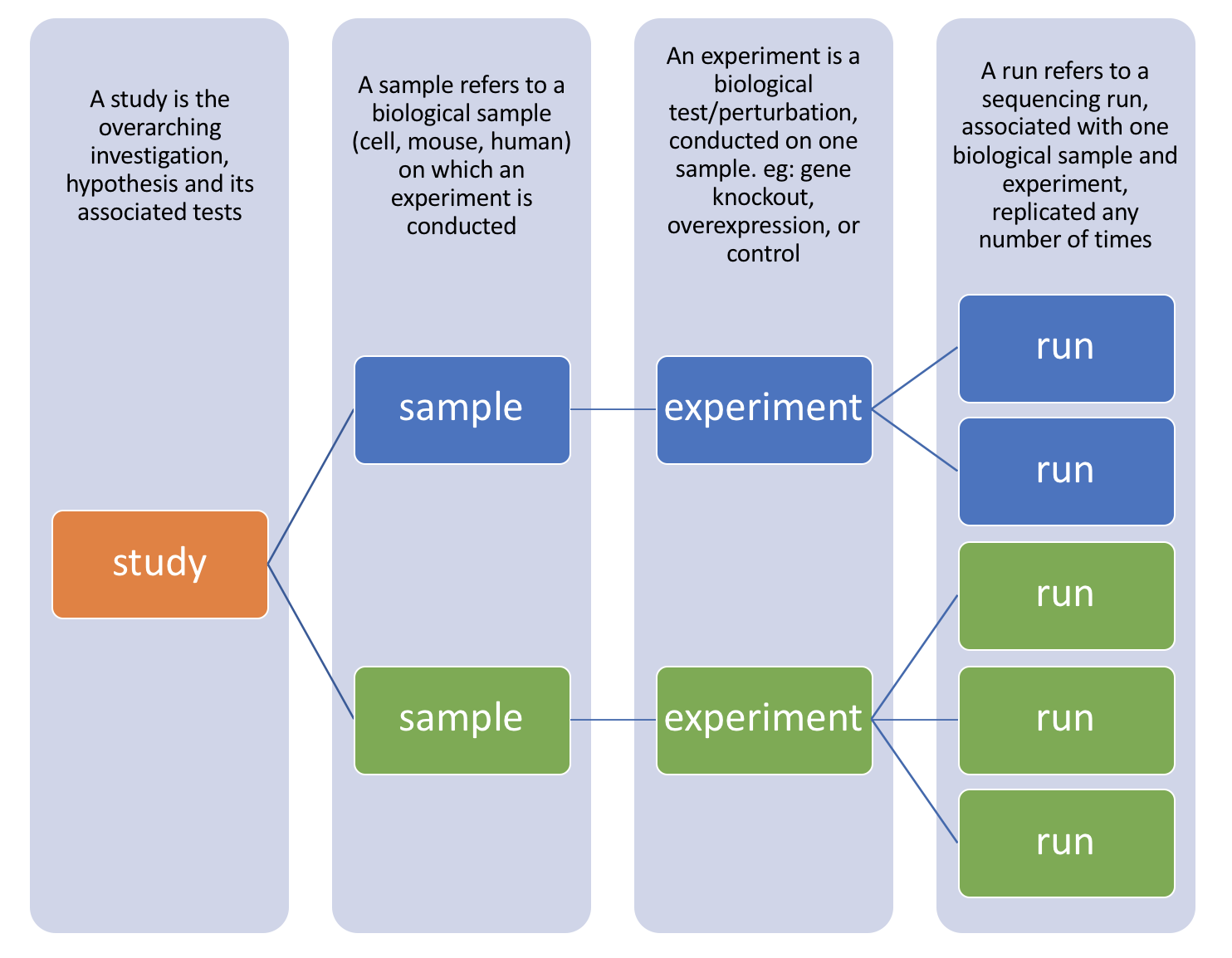
每個研究都會有個計畫編號(STUDY),開頭大多會是ERP或是SRP,這種計畫通常都會做多組檢體,而每個送去做定序的檢體都會有專屬編號(SAMPLE),以不同開頭表示不同意思/計畫,而此檢體使用的定序實驗設計,也會有個編號(EXPERIMENT),裡面會記錄此檢體所使用的定序機器種類、library type、定序設計(paired-end, single ….),而在同一次定序中其跑的run也會有編號,以SRR開頭。
PS. 一個包含多個的形式,例如一個study,可以代表多個sample。
檢索號(accession number) : 有各種前綴
STUDY :特定目的的研究課題或研究項目,可以包含多個研究機構和研究類型等, 和NCBI的BioProject資料庫裡面的項目相有關聯 。
study 包含了項目的所有metadata,並有一個NCBI 和EBI 共同承認的項目編號(universal project id)。
accession number通常以前綴 SRP,DRP,ERP 開頭(例如SRP000544)。
SAMPLE :樣本信息, 和NCBI的BioSample資料庫裡面的樣本相關聯,可以通過Trace來查詢。。
accession number通常以前綴 SRS,DRS,ERS 開頭(例如SRS001487) 。
EXPERIMENT : 實驗相關信息,一個實驗記載的實驗設計(Design),實驗平台(Platform)和結果處理(processing)三部分信息。
accession number通常以前綴 SRX,DRX,ERX 開頭。
RUN:RUN通俗理解就是定序上機運行一次產生的數據,是 SRA 裡面最小的概念,該編號通常直接連結到對應某一個/對fastq下機的文件。accession number 通常以前綴 SRR,DRR,ERR 開頭。
PS. 這裡提到另外一個概念 Submission
Submission:一個study 的項目整體數據,可以分多次遞交到SRA 資料庫。在一個項目啟動前期,就可以把study,experiment 的數據交上去,隨著項目的進展,一批一批提交 run 數據。
submission 等同於批次的概念。
到這邊發現好多前綴,一定很容易弄混
第一個字母:表示樣本最初被上傳到的來源資料庫
S – NCBI’s SRA database
E – EBI’s database
D – DDBJ database
第二個字母:固定為"R",代表Read
第三個字母:數據的類型,可以是項目、樣本、實驗或RUN
R – Run
X – Experiment
S – Sample
P – Project / study
01. Downloading SRA Toolkit · ncbi/sra-tools Wiki · GitHub
到這邊看作業系統,後下載~~~~
$ tar zxvf sratoolkit.XXX-ubuntu64.tar.gz
$ cd sratoolkit.XXX-ubuntu64
# 加入環境
$ echo 'export export PATH=$PATH:YOUR_PATH/sratoolkit.XXX-ubuntu64/bin' >> ~/.bash_profile
$ source ~/.bash_profile
好了之後可以嘗試在terminal 打
### 下載sra data to fastq 會用到的工具
prefetch -V
fastq-dump

可在上面搜索想要的檢體,例如我搜 HG005
在上面選擇想要的raw data,我做WGS ,因此我找一個 ILLUMINA (Illumina NovaSeq 6000)

找到了,看檔案大小100初G,是我要的平台機器跟WGS大小,因此選擇想要的。


我選擇 [SRR14724528](https://trace.ncbi.nlm.nih.gov/Traces?run=SRR14724528)
點入後有更多詳細的資訊

或是可以改用別的方式下載(AWS/GCP) / 直接下載 XD

prefetch --max-size 300G SRR14724528
因為想下載WGS ,檔案比較大,所以把prefetch原本限制無法下載超過20G的數據限制擴大。
SRA Run Selector 搜索 HG005 按

prefetch --option-file list.txt
fastq-dump vs fasterq-dump
簡單來說fasterq-dump是新版,比較快,但是缺點是沒辦法壓縮資料,所以之後我自己另外寫了一個壓縮的簡單script
常見參數--split-spot: 將pair end定序分為兩份,但是都放在同一個文件中 --split-files: 將pair end定序分為兩份,放在不同的文件,但是對於一方有而一方沒有的reads直接丟棄 --split-3 : 將pair end定序分為兩份,放在不同的文件,但是對於一方有而一方沒有的reads會單獨放在一個文件夾裡
-O : 輸出路徑
fasterq-dump SRR14724528 --split-3 -O ./sra_data

Accessing public genomic data: SRA | Accessing Public Genomic Data
HowTo: fasterq dump · ncbi/sra-tools Wiki · GitHub
https://www.jianshu.com/p/d1abdced8bcd


 iThome鐵人賽
iThome鐵人賽